| OS | Version | Chrome | Firefox | Microsoft Edge | Safari |
| Linux | Ubuntu 18.04 | 87.0.4280 | Not tested | n/a | n/a |
| MacOS | HighSierra | 87.0.4280 | 84.0 | n/a | 13.1.2 |
| Windows | 10 | 87.0.4280 | 83.0 | 87.0.664.66 | n/a |
ANNOTATION
The CRISPR-Cas systems are not only appealing systems to study adaptive bacterial and archaeal immunity against viral infection, but also have been extensively investigated due to their applications in the development of powerful genome editing tools. Meanwhile, viruses have evolved multiple anti-defense mechanisms during the host-parasite arms race to specifically inhibit CRISPR-Cas, such as diverse anti-CRISPR proteins (Acrs), which have enormous potential to be developed as modulators of genome editing tools.
CRISPRimmunity, an updated CRISPR-oriented web-tool, will dissect the key molecular events during the co-evolution of CRISPR and anti-CRISPR mechanisms, including CRISPR-Cas system detection and annotation, Anti-CRSISPR (Acr) protein and Acr-associated (Aca) gene annotation, self-targeting spacer search, repeat classification, prophage detection, bacteria-phage interaction detection based on spacer-protospacer matching, and integrated pipelines for novel Class 2 effector protein screening or Acr prediction.

Input
Users can upload a prokaryotic genome sequence file with any assembly level (complete, scaffold, contig and chromosome) , a genome file contains only one prokaryotic strain, the file format can be (Multi-)FASTA or (Multi-)GenBank format .

Parameter
8 modifiable parameters are supplied to Users corresponding the analysis module, 6 parameters are flexible, 2 parameters are fixed.
| Parameter definition | Default value | Corresponding module |
|---|---|---|
| The number of upstream or downstream proteins of the predicted CRISPR array | 10 | CRISPR-Cas System |
| The neighbour range of the predicted CRISPR array to identify signature genes | 20,000 bp | CRISPR-Cas Classification |
| The mismatch for results of identifying self-targeting | 2 | Self-targeting prediction |
| The coverage for results of identifying self-targeting, the interval of the value is from 0 to 1 | 1 | Self-targeting prediction |
| The mismatch for results of predicting interacting bacteriophages | 2 | Spacer targeting MGE detection |
| The coverage for results of predicting interacting bacteriophages, the interval of the value is from 0 to 1 | 0.9 | Spacer targeting MGE detection |
| The identity for results of predicting known Acr protein or Aca gene (fixed) | 1 | Known Acr protein and Aca gene annotation |
| The coverage for results of predicting known Acr protein or Aca gene (fixed) | 1 | Known Acr protein and Aca gene annotation |

Overview output

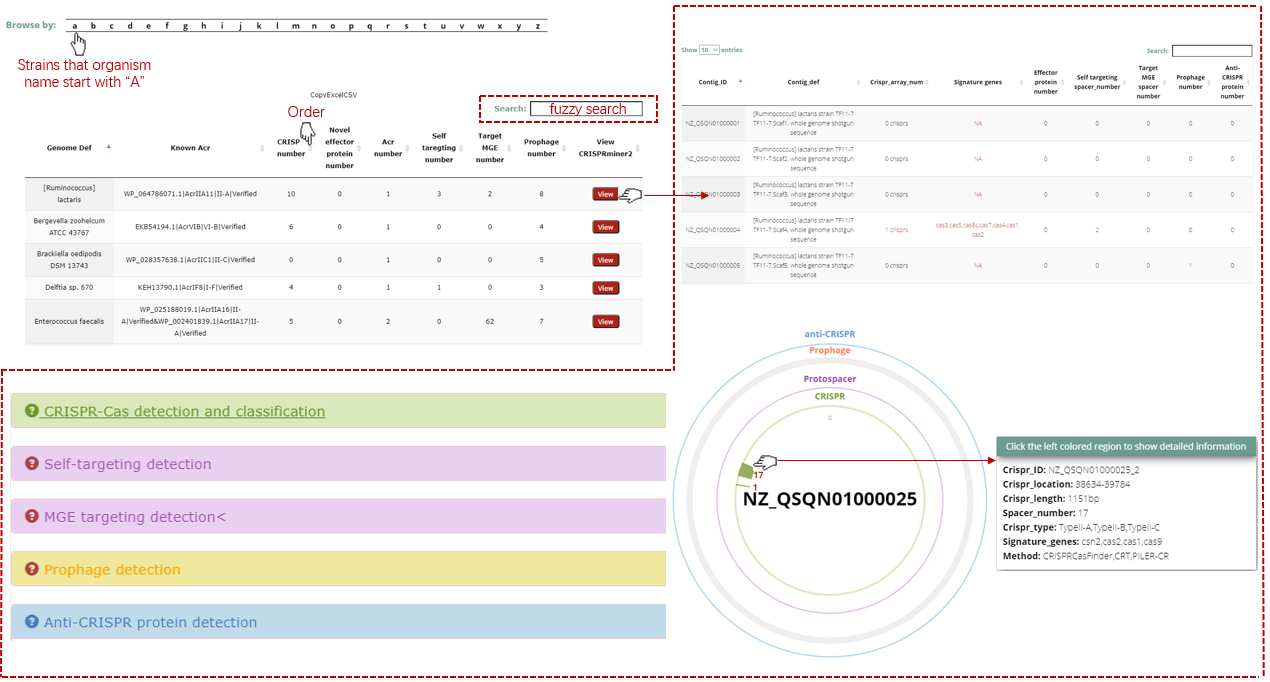
A table contains the detailed information of overview of prediction module and the description is shown in the following:
| Header | Description |
|---|---|
| Contig_ID | Accession ID of the input prokaryotic genome |
| Contig_def | Definition of the input prokaryotic genome |
| CRISPR array number | The number of CRISPR array detected in the prokaryotic genome |
| Signature genes | The Cas protein detected within 20kb of the CRISPR array |
| Self-targeting spacer number | The number of spacer targeting its own prokaryotic genome |
| MGE targeting spacer number | The number of spacer targeting the MGE genome included in our in-house MGE database |
| Prophage number | The number of prophage region in the prokaryotic genome |
| Anti-CRISPR protein number | The number of Anti-CRISPR protein verificated by experiment |
CRISPR-Cas Classification
CRISPRimmunity will provide the CRISPR-Cas system prediction results for the prokaryotic genome. A prokaryotic genome may contain both CRISPR array(s) and Cas genes, or CRISPR array(s) only, or Cas gene(s) only, or none of them. The (sub)type of identified CRISPR–Cas systems will be reported based on (sub)type signature Cas genes, the (sub)type of identified repeats will be reported based on repeat profile established by STSS. In addition to provide results in text, CRISPRimmunity provides visualization of the predicted systems.
(Sub)types of predicted CRISPR–Cas system
Two classes and six main types of the CRISPR—Cas system based on the defense mechanism & cas genes involved will be detected.
The class 1 system performs the function by a multi-subunit Cas protein complex, and the class 2 system requires only a single Cas protein (Cas9, Cas12 or Cas13) in the crRNA-effector complex.
(Sub)type can be identified by CRISPRimmunity:
| Class | Type | Subtype | Target | tracrRNA |
|---|---|---|---|---|
| 1 | I | I-A, I-B, I-C, I-D, I-E, I-F, I-U | DNA | No |
| 1 | III | III-A, III-B, III-C, III-D | DNA,RNA | No |
| 1 | IV | IV-A | DNA? | No |
| 2 | II | II-A, II-B, II-C | DNA | Yes |
| 2 | V | V-A(cas12a), V-B(cas12b), V-C(cas12c), V-D(casY), V-E(casX), V-F1(cas14a), V-F2(cas14b), V-G(cas12g), V-H(cas12h), V-I(cas12i), V-J(cas12j), V-K(cas12k), V-U(c2c4,c2c8,c2c10,c2c9), Cas14 family(c-k, U) | DNA,RNA(cas12g) | Partial Yes(V-B, V-E, V-F(cas14a), V-G, V-K) |
| 2 | VI | VI-A(cas13a), VI-B(cas13b), VI-C(cas13c), VI-D(cas13d) | RNA | No |



A table contains the detailed information of CRISPR-Cas system detection and annotation module and the description is shown in the following:
| Header | Description |
|---|---|
| CRISPR_ID | CRISPR locus ID of the input prokaryotic genome |
| CRISPR_location | CRISPR location of the current CRISPR locus |
| CRISPR_type | CRISPR type annotated by effector Cas protein(s) |
| Repeat_type | Repeat type annotated by sequence alignment with known type repeat database |
| Spacer_info | Spacer number of CRISPR array, detail spacer sequence can be obtained by click the icon |
| Cas protein info | Cas proteins within 20kbp of CRISPR array upstream and downstream |
| CRISPR-Cas_info | Click the Detail button to get the detailed information of CRISPR-Cas systems |
Self-targeting
Self-targeting is suggested as a form of autoimmunity[1], and has been used as a genomic marker to screen CRISPR-Cas inhibitor genes in Listeria monocytogenes[2]. CRISPRimmunity performs sequence alignment on its own genome with each spacer identified by CRISPR-Cas system module, the target (must outside bounds of found arrays) passing the screening criteria will be reported.

A table contains the detailed information of Self-targeting analysis module and the description is shown in the following:
| Header | Description |
|---|---|
| CRISPR_ID | CRISPR locus ID of the input prokaryotic genome. |
| Spacer_info | Spacer information of the current CRISPR locus, including CRISPR locus ID, spacer ID, spacer start location, spacer length, prokaryotic genome ID, method for predicting CRISPR array. |
| Spacer_region | The region of the spacer in the prokaryotic genome. |
| Spacer_length | The length of the spacer. |
| Hit_ID | The accession ID of the genome targeted by the spacer. |
| Protospacer_location | The location of the genome targeted by the spacer. |
| Mismatch | The mismatch base number of the sequence between spacer and protospacer. |
| Identity | The identity of the sequence between spacer and protospacer. The interval of the value is from 0 to 1. |
Targeting for MGE
Phage-host interactions are appealing systems to study bacterial adaptive evolution and are increasingly recognized as playing an important role in human health and diseases, which may contribute to novel therapeutic agents, such as phage therapy to combat multi-drug resistant infections. CRISPRimmunity performs sequence alignment on phage genome within the pre-established phage database with each spacer identified by CRISPR-Cas system module, the target passing the screening criteria will be reported. The phage genome database contained 10,463 complete phage genome sequences collected from Millardlab[3], which were extracted from the GenBank database on May 2019.

A table contains the detailed information of Bacteria-phage interaction detection module and the description is shown in the following:
| Header | Description |
|---|---|
| CRISPR_ID | CRISPR locus ID of the input prokaryotic genome |
| Spacer_info | Spacer information of the current CRISPR locus, including CRISPR locus ID, spacer ID, spacer start location, spacer length, prokaryotic genome ID, method for predicting CRISPR array. |
| Spacer_region | The region of the spacer in the prokaryotic genome. |
| Spacer_length | The length of the spacer. |
| Hit_phage_ID | The accession ID of the phage genome targeted by the spacer. |
| Hit_phage_def | The definition of the phage genome targeted by the spacer. |
| Protospacer_location | The location of the genome targeted by the spacer. |
| Mismatch | The mismatch base number of the sequence between spacer and protospacer. |
| Identity | The identity of the sequence between spacer and protospacer. The interval of the value is from 0 to 1. |
Prophage regions
Identifying relationship between bacteria and phage is based on the phenomenon that many phages insert their genomes into that of their hosts, the integrated phages are known as prophages. There are two key steps for prediction using prophage analysis. The first step is to identify the prophage region in the bacteria genome, and the second step is to annotate the prophage region using BLASTN or BLASTP method by checking the similarity of DNA or protein sequence between the prophage region and Uniprot virus genomes.
DBSCAN-SWA detects prophage by combining DBSCAN and Sliding Window Algorithm.It uses density-based spatial clustering of applications with noise (DBSCAN) [4] to predict clusters of phage or phage-like genes and uses sliding window algorithm to predict these regions based on these bacterium proteins with enough proteins that are not predicted in DBSCAN. More detail information about the DBSCAN-SWA could be seen in the article [5].

A table contains the detailed information of Prophage detection module and the description is shown in the following:
| Header | Description |
|---|---|
| Region | The index of the prophage region. |
| Region Position | The position of the prophage region. |
| Protein_number | The number of proteins within the prophage region. |
| Hit_taxonomy | Genus of the most like phage integrated into the prokaryotic genome, the probability values are marked in brackets. |
| Key_proteins | Key phage-related proteins |
| Att_site | The information of the attL and attR, the value is set NA if attL/attR can not be detected. |
| Prophage annotation | Click the Detail button to get the detailed information of prophage region. |
A table contains the detailed information of prophage region and the description is shown in the following:
| Header | Description |
|---|---|
| Protein_ID | Accession ID of the protein within the prophage region. |
| Protein_Def | Definition of the protein within the prophage region. |
| Hit_ID | Homologous protein in UniProt database. |
| Hit_Def | The organism of the homologous protein. |
| E-value | The E-value of the protein sequence alignment. |
| Identity | The identity of the protein sequence alignment. The interval of the value is from 0 to 1. |
Novel Anti-CRISPRs
Three strategy are used for discovering the inhibitor of the CRISPR-Cas system. One strategy is to identify the homologous proteins of the known Anti-CRISPR proteins. Identify the novel Anti-CRISPR protein based on Acr-associated protein is another strategy. An alternative inhibitor discovery strategy, the presence of stable self-targeting CRISPR sequences is applied as a potential indicator of genomes or mobile genetic elements (MGEs) harboring CRISPR inhibitors[6-7].

A table contains the detailed information of Anti-CRISPR and Acr-associated (Aca) protein detection module and the description is shown in the following:
| Header | Description |
|---|---|
| Acr_ID | Accession ID of the Anti-CRISPR protein candidate |
| Acr_info | The information of the Anti-CRISPR protein candidate |
| Acr size | The size of the Anti-CRISPR protein candidate |
| Homology with known anti | The homologous known Anti-CRISPR protein of the Anti-CRISPR protein candidate |
| Neighbor Aca | The information of the Aca in the neighbor of novel Anti-CRISPR protein candidate |
| in_prophage | The location of the prophage region containing the novel Anti-CRISPR protein candidate |
| ST in prophage | The number of self-targeting in prophage region |
| Neighbor HTH | The information of the HTH in the neighbor of novel Anti-CRISPR protein candidate |
| Predict by AcRanker | Whether the the Anti-CRISPR protein candidate be predicted by AcRanker |
PREDICTION
Anti-CRISPRs
Three strategy are used for discovering the inhibitor of the CRISPR-Cas system. One strategy is to identify the homologous proteins of the known Anti-CRISPR proteins. Identify the novel Anti-CRISPR protein based on Acr-associated protein is another strategy. An alternative inhibitor discovery strategy, the presence of stable self-targeting CRISPR sequences is applied as a potential indicator of genomes or mobile genetic elements (MGEs) harboring CRISPR inhibitors[6-7].

Input

3 modifiable parameters are supplied to Users corresponding the analysis module.
| Parameter definition | Default value |
|---|---|
| The identity for results of predicting homologs of known anti-CRISPR protein, the interval of the value is from 0 to 1. | 0.4 |
| The coverage for results of predicting homologs of known anti-CRISPR protein, the interval of the value is from 0 to 1. | 0.7 |
| The number of upstream and downstream proteins for predicting novel anti-CRISPR protein by applying "guilt by association". | 3 |

Output

CRISPR-Cas system detection and annotation
CRISPRimmunity predicts novel class 2 CRISPR-Cas loci using comparative genomics screening approach as displayed in the following flowchart

Input
Users can upload or paste a prokaryotic genome sequence file with any assembly level (complete, scaffold, contig and chromosome) , a genome file contains only one prokaryotic strain, the file format can be (Multi-)FASTA or (Multi-)GenBank format .

4 modifiable parameters are supplied to Users corresponding the analysis module.
| Parameter definition | Default value |
|---|---|
| The minimal protein size of the novel effector protein. | 500aa |
| The number of proteins distant from CRISPR array to predict novel effector protein. | 5 |
| The identity for finding homolog proteins of candidate novel effector proteins, the interval of the value is from 0 to 1. | 0.3 |
| The coverage for finding homolog proteins of candidate novel effector proteins, the interval of the value is from 0 to 1. | 0.6 |

Output




CASE STUDY
Known Acr-associated (Aca) protein
We collected 99 known Anti-CRISPR proteins from anti-CRISPRdb. These bacterial genomes were analysed using the integrated programs of CRISPRimmunity including CRISPR-Cas, novel effector protein identification, Self-Targeting, bacterial-phage interaction, prophage and Anti-CRISPR proteins.
Supplementary
File format
FASTA format
Definiton
FASTA format is a text-based format for representing either nucleotide sequences or peptide sequences, in which nucleotides or amino acids are represented using single-letter codes[8-9]. A FASTA format sequence starts with a single comment line and is followed by sequence lines. A greater-than (“>”) symbol is used before the first character of the comment line to distinguish it from sequence lines.
Example
>sequenceID-001 description
AAGTAGGAATAATATCTTATCATTATAGATAAAAACCTTCTGAATTTGCTTAGTGTGTAT
ACGACTAGACATATATCAGCTCGCCGATTATTTGGATTATTCCCTG
A multi-FASTA file contains multiple FASTA formated sequences.
Example
>sequenceID-001 description
AAGTAGGAATAATATCTTATCATTATAGATAAAAACCTTCTGAATTTGCTTAGTGTGTAT
ACGACTAGACATATATCAGCTCGCCGATTATTTGGATTATTCCCTG
>sequenceID-002 description
CAGTAAAGAGTGGATGTAAGAACCGTCCGATCTACCAGATGTGATAGAGGTTGCCAGTAC
AAAAATTGCATAATAATTGATTAATCCTTTAATATTGTTTAGAATATATCCGTCAGATAA
TCCTAAAAATAACGATATGATGGCGGAAATCGTC
>sequenceID-003 description
CTTCAATTACCCTGCTGACGCGAGATACCTTATGCATCGAAGGTAAAGCGATGAATTTAT
CCAAGGTTTTAATTTG
GenBank(full) format
Definiton
The GenBank(full) format file contains many biological features of the record, including Locus line, sequence length, molecule type, record definition, accession id and others. The GenBank(full) format (GenBank Flat File Format) consists of an annotation section and a sequence section. The start of the annotation section is marked by a line beginning with the word “LOCUS”. The start of sequence section is marked by a line beginning with the word “ORIGIN” and the end of the section is marked by a line with only “//“.
Example
LOCUS EU490707 1302 bp DNA linear PLN 05-MAY-2008
DEFINITION Selenipedium aequinoctiale maturase K (matK) gene, partial cds;
chloroplast.
ACCESSION EU490707
VERSION EU490707.1 GI:186972394
KEYWORDS .
SOURCE chloroplast Selenipedium aequinoctiale
ORGANISM Selenipedium aequinoctiale
Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta;
Spermatophyta; Magnoliophyta; Liliopsida; Asparagales; Orchidaceae;
Cypripedioideae; Selenipedium.
REFERENCE 1 (bases 1 to 1302)
AUTHORS Neubig,K.M., Whitten,W.M., Carlsward,B.S., Blanco,M.A.,
Endara,C.L., Williams,N.H. and Moore,M.J.
TITLE Phylogenetic utility of ycf1 in orchids
JOURNAL Unpublished
REFERENCE 2 (bases 1 to 1302)
AUTHORS Neubig,K.M., Whitten,W.M., Carlsward,B.S., Blanco,M.A.,
Endara,C.L., Williams,N.H. and Moore,M.J.
TITLE Direct Submission
JOURNAL Submitted (14-FEB-2008) Department of Botany, University of
Florida, 220 Bartram Hall, Gainesville, FL 32611-8526, USA
FEATURES Location/Qualifiers
source 1..1302
/organism="Selenipedium aequinoctiale"
/organelle="plastid:chloroplast"
/mol_type="genomic DNA"
/specimen_voucher="FLAS:Blanco 2475"
/db_xref="taxon:256374"
gene <1..>1302
/gene="matK"
CDS <1..>1302
/gene="matK"
/codon_start=1
/transl_table=11
/product="maturase K"
/protein_id="ACC99456.1"
/db_xref="GI:186972395"
/translation="IFYEPVEIFGYDNKSSLVLVKRLITRMYQQNFLISSVNDSNQKG
FWGHKHFFSSHFSSQMVSEGFGVILEIPFSSQLVSSLEEKKIPKYQNLRSIHSIFPFL
EDKFLHLNYVSDLLIPHPIHLEILVQILQCRIKDVPSLHLLRLLFHEYHNLNSLITSK
KFIYAFSKRKKRFLWLLYNSYVYECEYLFQFLRKQSSYLRSTSSGVFLERTHLYVKIE
HLLVVCCNSFQRILCFLKDPFMHYVRYQGKAILASKGTLILMKKWKFHLVNFWQSYFH
FWSQPYRIHIKQLSNYSFSFLGYFSSVLENHLVVRNQMLENSFIINLLTKKFDTIAPV
ISLIGSLSKAQFCTVLGHPISKPIWTDFSDSDILDRFCRICRNLCRYHSGSSKKQVLY
RIKYILRLSCARTLARKHKSTVRTFMRRLGSGLLEEFFMEEE"
ORIGIN
1 attttttacg aacctgtgga aatttttggt tatgacaata aatctagttt agtacttgtg
61 aaacgtttaa ttactcgaat gtatcaacag aattttttga tttcttcggt taatgattct
121 aaccaaaaag gattttgggg gcacaagcat tttttttctt ctcatttttc ttctcaaatg
181 gtatcagaag gttttggagt cattctggaa attccattct cgtcgcaatt agtatcttct
241 cttgaagaaa aaaaaatacc aaaatatcag aatttacgat ctattcattc aatatttccc
301 tttttagaag acaaattttt acatttgaat tatgtgtcag atctactaat accccatccc
361 atccatctgg aaatcttggt tcaaatcctt caatgccgga tcaaggatgt tccttctttg
421 catttattgc gattgctttt ccacgaatat cataatttga atagtctcat tacttcaaag
481 aaattcattt acgccttttc aaaaagaaag aaaagattcc tttggttact atataattct
541 tatgtatatg aatgcgaata tctattccag tttcttcgta aacagtcttc ttatttacga
601 tcaacatctt ctggagtctt tcttgagcga acacatttat atgtaaaaat agaacatctt
661 ctagtagtgt gttgtaattc ttttcagagg atcctatgct ttctcaagga tcctttcatg
721 cattatgttc gatatcaagg aaaagcaatt ctggcttcaa agggaactct tattctgatg
781 aagaaatgga aatttcatct tgtgaatttt tggcaatctt attttcactt ttggtctcaa
841 ccgtatagga ttcatataaa gcaattatcc aactattcct tctcttttct ggggtatttt
901 tcaagtgtac tagaaaatca tttggtagta agaaatcaaa tgctagagaa ttcatttata
961 ataaatcttc tgactaagaa attcgatacc atagccccag ttatttctct tattggatca
1021 ttgtcgaaag ctcaattttg tactgtattg ggtcatccta ttagtaaacc gatctggacc
1081 gatttctcgg attctgatat tcttgatcga ttttgccgga tatgtagaaa tctttgtcgt
1141 tatcacagcg gatcctcaaa aaaacaggtt ttgtatcgta taaaatatat acttcgactt
1201 tcgtgtgcta gaactttggc acggaaacat aaaagtacag tacgcacttt tatgcgaaga
1261 ttaggttcgg gattattaga agaattcttt atggaagaag aa
//
A multi-GenBank(full) file contains multiple GenBank(full) formated sequences.
Repeat file format
Definition
Repeat sequence can be obtain from result of the CRISPR-Cas association tools, such as CRISPRCasfinder, PILERCR, CRT. The input file for the repeat type annotation is organizing the repeat to (multi)FASTA format
Example
>NC_003155_11_CRT
CGACCCACCTCCGCTCGCGCGGAGAGAAC
>NC_003155_11_PILER-CR
CGACCCACCTCCGCTCGCGCGGAGAG
>NC_003155_12_PILER-CR
CGGTTCACCTCCGCTCGCGCGGAGAGCAC
>NC_003155_12_CRISPRCasFinder
GTGCTCTCCGCGCGAGCGGAGGTGAACCG
Reference
[1] Stern, A., Keren, L., Wurtzel, O., Amitai, G. & Sorek, R. Self-targeting by CRISPR: gene regulation or autoimmunity? Trends Genet. 26, 335–340 (2010).
[2] Bondy-Denomy J. Protein Inhibitors of CRISPR-Cas9. ACS Chem Biol. 2018 Feb 16;13(2):417-423. doi: 10.1021/acschembio.7b00831. Epub 2018 Jan 17. PMID: 29251498; PMCID: PMC6049822.
[3] Ester,M., Kriegel,H.P., Sander,J. and Xu,X. (1996) A density-based algorithm for discovering clusters in large spatial databases with noise. KDD-1996 Proceedings. AAAI Press, Menlo Park, CA, pp. 226–231.
[4] You Zhou, Yongjie Liang, Karlene H. Lynch, Jonathan J. Dennis, David S. Wishart (2010) "PHAST: A Fast Phage Search Tool" (Nucleic Acid Research submitted)
[5] Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC, Hugenholtz P. CRISPR Recognition Tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics. 2007 Jun 18;8(1):209 [6]Rauch BJ, Silvis MR, Hultquist JF, Waters CS, McGregor MJ, Krogan NJ, Bondy-Denomy J. Inhibition of CRISPR-Cas9 with Bacteriophage Proteins. Cell. 2017 Jan 12;168(1-2):150-158.e10. doi: 10.1016/j.cell.2016.12.009. Epub 2016 Dec 29. PMID: 28041849; PMCID: PMC5235966.
[7] http://millardlab.org/bioinformatics/bacteriophage-genomes/